
|
E. Prabhu Raman
-
Postdoctoral Fellow
Dept. of Pharmaceutical Sciences
University of Maryland, Baltimore
CV,
Google Scholar,
PubMed
- Email:
-
|
Education
- Ph.D Computational Biophysics, 2008
- M.Sc. (Hons.) Mathematics, 2002
Current Research
- Computational Study of Molecular Recognition
- Computer Aided Drug Design Methods
- Molecular Mechanics Force Field Development
- Cheminformatics
|
My current research efforts are centred on understanding non covalent molecular recognition and developing computational methods to model it. Molecular recognition forms an invisible wiring diagram forming the physical basis of many biological functions. Therefore its accurate and efficient modeling offers immense potential for the discovery of therapeutics. Of particular interest is protein-ligand interaction prediction, which has applications in small molecule drug design and discovery. My primary research tools are atomic detail simulations and classical molecular mechanics models which offer a system independent approach to compute thermodynamic and kinetic properties. While statistical thermodynamics rigorously connects molecular microstates to macroscopic properties including binding free energies, there exist significant hurdles in efficiently calculating those properties. Prominent challenges include the sampling of the large conformational spaces of the protein, ligand and solvent, and estimating macroscopic properties from the samples. The research projects described below highlight my efforts towards some of those problems.
Molecular Driving Forces Implicated in Protein-Ligand Binding
While the protein-ligand binding problem has been theoretically well formulated in the statistical thermodynamics context, quantitative accounting of the contributions of solvent, and other degrees of freedom is largely lacking. In a recent contribution, we developed a rigorous approach to calculate the enthalpic and entropic contributions of the ligand and solvent degrees of freedom to binding thermodynamics. Using this method, we analyzed the binding of small fragment-sized non-polar and polar ligands to tens of different binding pockets on proteins. Our analysis provided detailed mechanistic insights into binding and the hydrophobic effect. The method also opens up exciting possibilities for calculating absolute and relative binding affinities, and avenues for developing approximations to model binding efficiently.
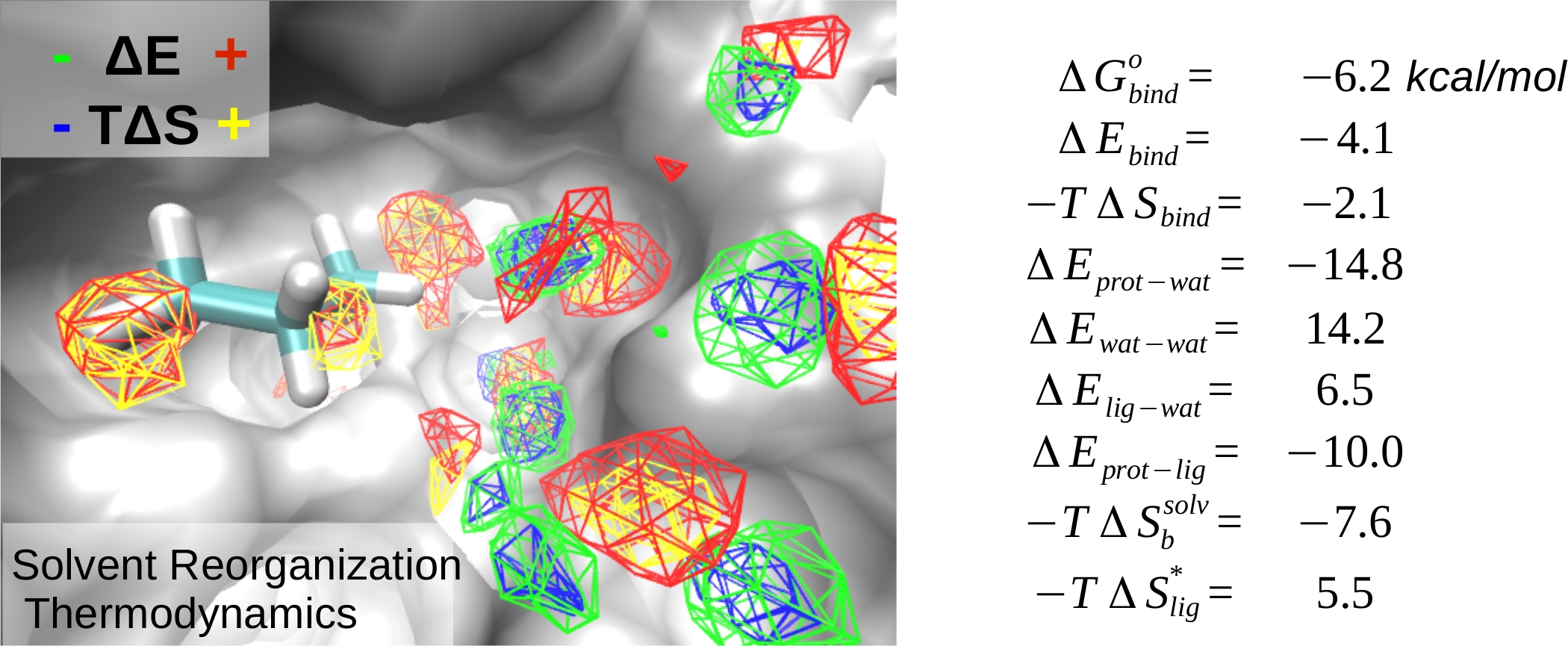
Raman EP and Mackerell AD Jr., Spatial Analysis and Quantification of the Thermodynamic Driving Forces in Protein-Ligand Binding: Binding Site Variability, J. Am. Chem. Soc. 2015 (in-press)
[PubMed Central]
Hydration Thermodynamics in Biomolecular Interfaces
Water plays a dominant role in non-covalent binding, but rigorous and efficient methods to calculate its effect have been elusive. We developed an efficient method for sampling of water configurations and estimation of thermodynamics. Grand canonical Monte Carlo (GCMC) sampling implemented in a grid-based representation of proteins was combined with the grid inhomogeous solvation theory method (GIST) to estimate excess thermodynamic properties in binding sites. The method matches the accuracy of free energy perturbation (FEP), which suggests promising prospects for the method in estimating water reorganization thermodynamics as a result of ligand binding.
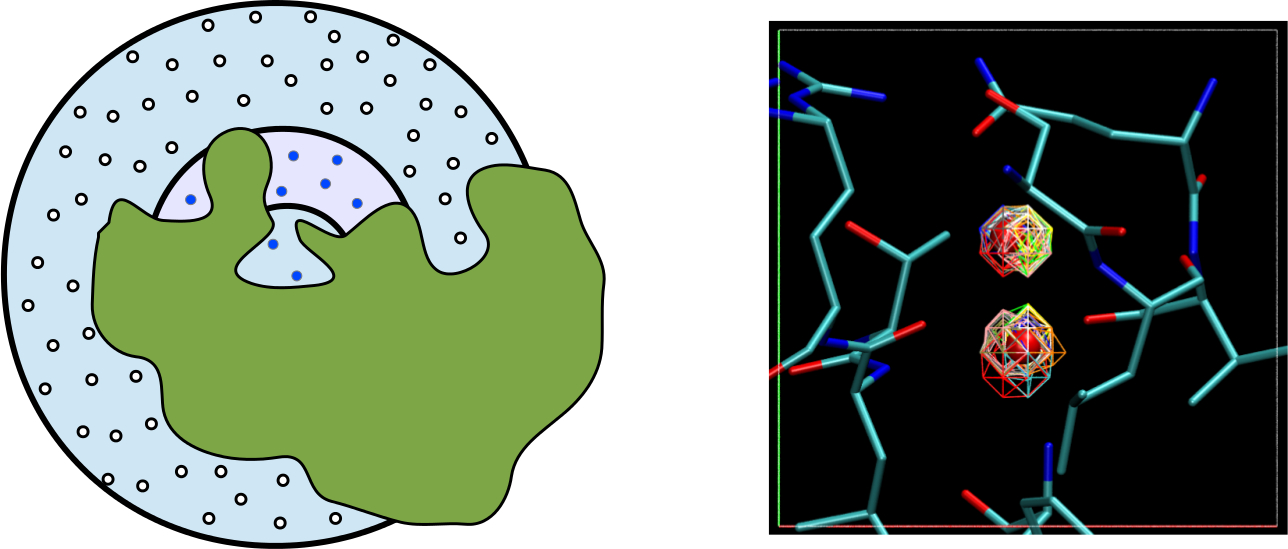
Raman EP and Mackerell AD Jr., Rapid estimation of hydration thermodynamics of
macromolecular regions, J. Chem. Phys. 2013, 139(6), 055105
[PubMed Central]
|

|
Computing Protein Affinity Patterns for Drug Design The design of low molecular weight ligands that can bind to a protein of determined structure with high affinity is a central problem in drug discovery. Proteins can adopt diverse shapes when binding to different ligands, posing a significant hurdle in the computational discovery or design of ligands. A second problem is posed by the astronomical chemical space of drug-like molecules, making it impossible to screen all possible ligands to find the most optimal ones. An apt analogy of the problem is finding a needle in not one, but a million haystacks! As an effective solution to this dual problem, we have developed and applied the Site Identification by Ligand Competitive Saturation (SILCS) method. The method involves MD simulations of proteins in an aqueous solution of small molecular fragments to map the affinity pattern of the protein for diverse functional groups while including protein flexibility and explicit solvation. A grid based free energy map of fragment binding is computed, which can be used qualitatively and quantitatively in early as well as late stages of drug design. We showed the ability of the method to identify locations of ligand functional groups in crystal structures, and developed methods for scoring arbitrary ligands for use in virtual screening and lead optimization. See the publications below for additional applications. Also see http://silcsbio.com
|
Raman EP, Yu W, Guvench O, Mackerell AD Jr., Reproducing crystal binding modes of ligand functional groups using Site-Identification by Ligand Competitive Saturation (SILCS) simulations, J. Chem. Inf. Model. 2011, 51(4), 877
[PubMed Central]
Raman EP, Yu W, Lakkaraju SK, Mackerell AD Jr., Inclusion of multiple fragment types in the site identification by ligand competitive saturation (SILCS) approach, J. Chem. Inf. Model. 2013, 53(12), 3384
Lakkaraju SK, Raman EP, Yu W, Mackerell AD Jr., Sampling of Organic Solutes in Aqueous and Heterogeneous Environments Using Oscillating Excess Chemical Potentials in Grand Canonical-like Monte Carlo-Molecular Dynamics Simulations, J. Chem. Theor. Comput. 2014, 10(6), 2281
Yu W, Lakkaraju SK, Raman EP, Mackerell AD Jr., Site-Identification by Ligand Competitive Saturation (SILCS) assisted pharmacophore modeling, J. Comput. Aided Mol. Des. 2014, 28(5), 491
Yu W, Lakkaraju SK, Raman EP, Fang L, Mackerell AD Jr., Pharmacophore modeling using Site-Identification by Ligand Competitive Saturation (SILCS) with multiple probe molecules, J. Chem. Inf. Model. 2015 (in-press)
Rapid Calculation of Relative Binding Free Energies
Lead optimization is an expensive and time consuming stage in drug design, where lead compounds are modified to improve potency and drug-like properties. In this stage, the free energy perturbation (FEP) method has been found to be very useful as one can predict the consequence of the modification on binding affinity. We developed a protocol to implement small perturbations in a single step from the conformational ensemble of the lead ligand. The protocol when applied to an ensemble of SILCS simulations is able to efficiently identify a range of fragments that can bind protein pockets with higher affinity than the explicitly simulated fragments. Secondly, the method is able to rapidly screen hundreds of possibilities of small compound modifications for affinity enhancement, facilitating lead optimization.
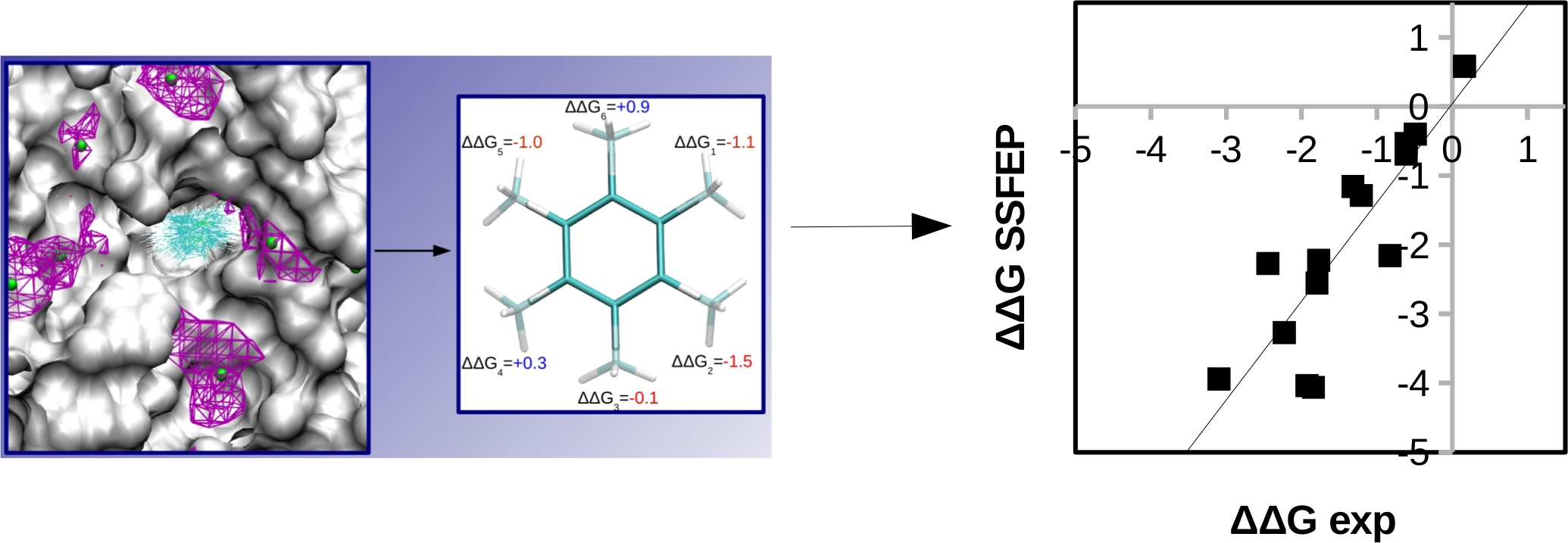
Raman EP, Vanommeslaeghe K, MacKerell AD Jr., Site-Specific fragment identification guided by single-step free energy perturbation calculations, J. Chem. Theor. Comput. 2012, 8(10), 3513
[PubMed Central]
Molecular Mechanics Force Field Development
Classical molecular mechanics (MM) presents an effective approximation that has enabled studies of a wide range of molecules in biological and chemical applications. However, the accuracy of this approach is crucially dependent on the quality of force-field parameters. I have been a co-developer of the CHARMM carbohydrate and general force-fields, where the parametrization strategy entails a self-consistent approach to reproduce both theoretical (Quantum Mechanical, QM) and experimental data as shown below.
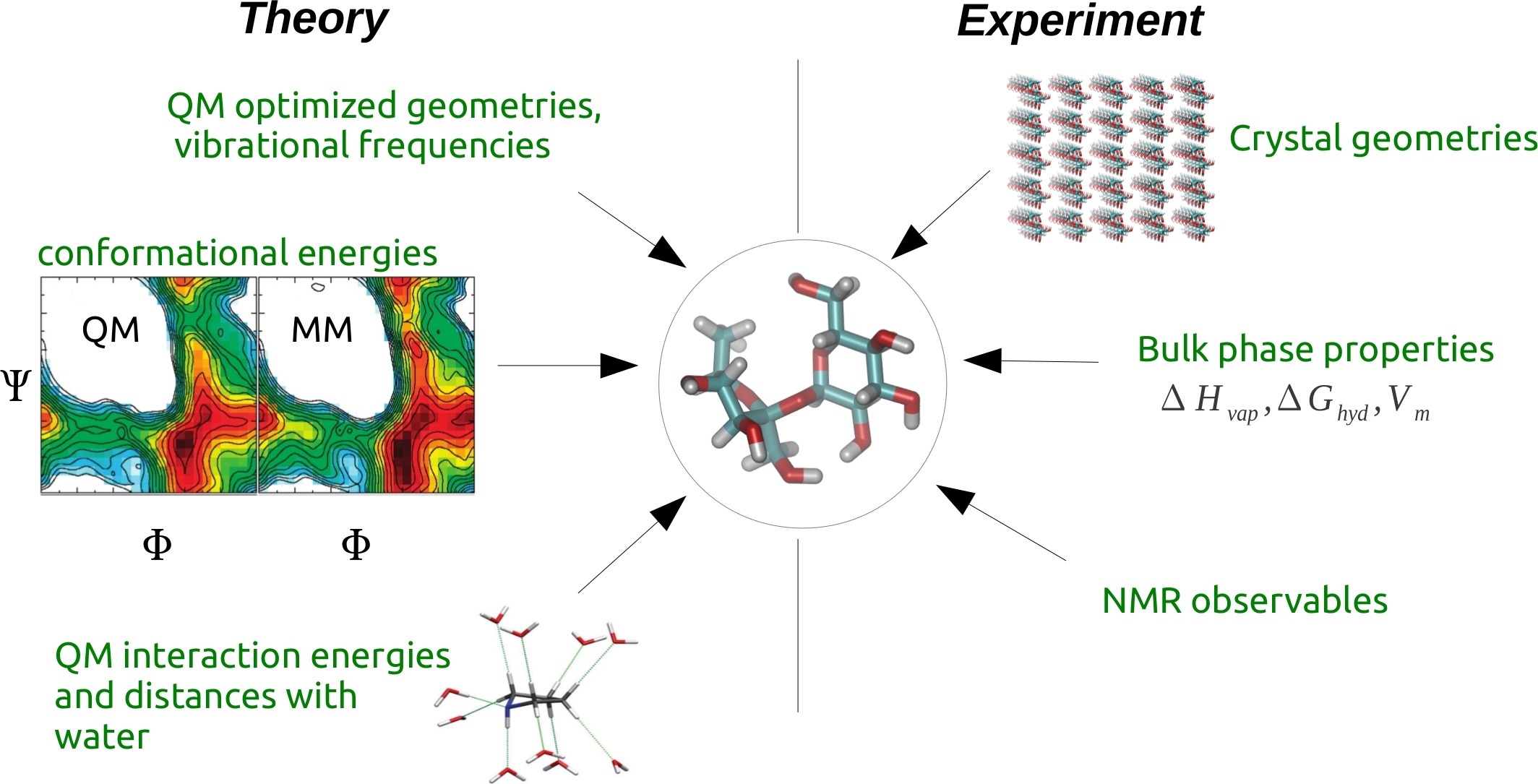
Raman EP, Guvench O, Mackerell AD Jr., CHARMM additive all-atom force field for glycosidic linkages in carbohydrates involving furanoses, J. Phys. Chem. B 2010, 114(40), 12981
[PubMed Central]
Abel S, Dupradeau FY, Raman EP, MacKerell AD Jr., Marchi M. Molecular simulations of dodecyl-beta-maltoside micelles in water: influence of the headgroup conformation and force field parameters, J. Phys. Chem. B 2011, 115(3), 487
[PubMed Central]
Guvench O, Mallajosyula SS, Raman EP, Hatcher E, Vanommeslaeghe K, Foster TJ, Jamison FW 2nd, Mackerell AD Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling, J. Chem. Theor. Comput. 2011, 7(10), 3162
[PubMed Central]
Vanommeslaeghe K, Raman EP, MacKerell AD Jr., Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges, J. Chem. Inf. Model. 2012, 52(12), 3155
[PubMed Central]
Covers
