|
Research in the MacKerell lab involves the development and application of computational methods to investigate the relationships of structure and dynamics to function in a range of biological and chemical systems. These efforts range from empirical force field development, including the CHARMM36 and classical Drude polarizable force fields, development of novel solute and conformational sampling methodologies, understanding the physical forces driving the structure and dynamics of proteins, nucleic acids and carbohydrates and computer-aided drug design (CADD) studies including development of novel methods such as the Site Identification by Ligand Compepetive Saturation approach. A number of recent publications in these areas are highlighted below with more information available on the Research page and from our list of publications. In addition, force fields and other utilities developed in the lab may be accessed via the CHARMM page. Research Highlight: RNA simulations with the Drude Polarizable Force Field: MD simulations of RNAs have traditionally been challenging with additive force fields, especially in non-canonical regions such as in RNA hairpins. In a recent study using an updated version of the polarizable Drude-2017 nucleic acid force field [DOI] it was shown that significant improvements in stability were acheived associated with better reproduction of non-canonical base-base interactions. Shown below are images of the crystal structures of the studied hairpins along with structures obtained from MD simulations using the Drude and C36 force fields. See: Sengul, M.Y. and MacKerell, A.D., Jr., "Accurate modeling of RNA hairpins through the explicit treatment of electronic polarizability with the classical Drude oscillator force field," Journal of Computational Biophysics and Chemistry, 21: 1-11, 2022, PMC9231828, [DOI]. Please contact us for the version of the Drude force field used in that study. 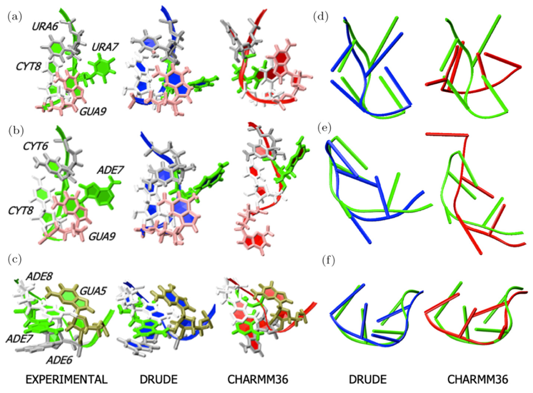
Research Highlight: Drude General Force Field (DGenFF): Towards the use of the polarizable Drude force field for drug-like molecules we've made advances in the coverage of chemical space and in the automated generation of electrostatic parameters. To facilitate coverage of chemical space a deep neural net (DNN) approach was developed to more comprehensively sample Lennard-Jones parameters for new atom types. See Chatterjee et al., "Harnessing Deep Learning for Optimization of Lennard-Jones Parameters for the Polarizable Classical Drude Oscillator Force Field, Journal of Chemical Theory and Computation, 18: 2388-2407, 2022, PMC9097857, [DOI]. To automatically generate partial atomic charges, atomic polarizabilities (alphas), and atomic Thole scale factors for organic molecules data for close to 40,000 compounds were generated using quantum mechanical calculations. That data was then used to train DNNs to predict the electrostatic parameters (TOC graphic below). For details see Kumar et al., "Deep Neural Network Model to Predict the Electrostatic Parameters in the Polarizable Classical Drude Oscillator Force Field," Journal of Chemical Theory and Computation, 18:1711-1725, 2022, PMC8904317, [DOI]. In addition, a parameter optimization GUI, FFParam, is now available to facilitate small molecule parameter optimization on the context of both the C36/CGenFF additive and Drude polarizable force fields.Kumar et al., "FFParam: Standalone Package for CHARMM Additive and Drude Polarizable Force Field Parametrization of Small Molecules," Journal of Computational Chemistry, 41: 958-970, 2020, PMC7323454, [DOI]. It may be downloaded from our website force field resouces. And to facilitate use of the Drude force field a Drude Prepper module has been added to the CHARMM-Gui as described in Kognole et al., "CHARMM-GUI Drude Prepper for Molecular Dynamics Simulation Using the Classical Drude Polarizable Force Field" Journal of Computational Chemistry, 43:359-375, 2022, PMC8741736, [DOI]. 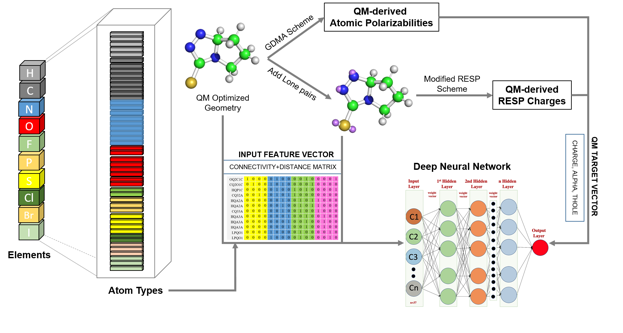
Research Highlight: SILCS: We continue to implement methods associated with the Site Identification by Ligand Competitive Saturation (SILCS) technology as well as apply it to interesting biological problems. SILCS has been extended for use in small-molecule drug design targeting RNA as reported in Kognole, A.A., Hazel, A., and MacKerell, A.D., Jr., "SILCS-RNA: Toward a Structure-Based Drug Design Approach for Targeting RNAs with Small Molecules," Journal of Chemical Theory and Computation, 18: 5672-5691, 2022, PMC9474704, 2022, [DOI]. The approach has also been extended for use in the formulation of protein biologics, including monoclonal antibodies as descibed in Jo et al. "Computational Characterization of Antibody-Excipient Interactions for Rational Excipient Selection using the Site Identification by Ligand Competitive Saturation (SILCS)-Biologics Approach," Molecular Pharmaceutics, 17: 4323-4333, 2020, PMC7606568 [DOI]. In an interesting application study, the SILCS-Hotspots and Pharmacophore approaches were used to identify a novel allosteric binding sites and allosteric modulators targeting that site in the b2-adrenergic receptors, a GPCR. See image below and details in Sushrut et al., "In silico identification of a beta2-adrenoceptor allosteric site that selectively augments canonical Beta2AR-Gs signaling and function," Proceedings of the National Academies of Sciences, USA, 119: e2214024119, 2022, PMC9894167, [DOI] 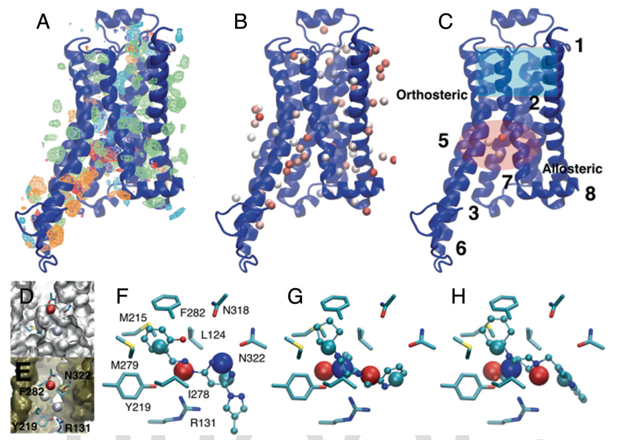
To explore more about who we are and what we do, use the navigation links above. |
 |