CGenFF
This page was originally developed by Kenno Vanommeslaeghe here.
University of Maryland, Baltimore
Baltimore, MD 21201
USA
|
|
CGenFFThis page was originally developed by Kenno Vanommeslaeghe here. |
University of Maryland, Baltimore |
./install.com gnu medium +CGENFFrm -r build/gnu lib/gnu
* check whether CHARMM was built with CGenFF support
*
echo ?CGENFF
RDCMND: can not substitute energy "?CGENFF"read rtf card name @TOPPAR/top_all36_prot.rtf
read param card flex name @TOPPAR/par_all36_prot.prm
read rtf card append name @TOPPAR/top_all36_cgenff.rtf
read para card flex append name @TOPPAR/par_all36_cgenff.prm
stream toppar_water_ions.str
stream my_ligands.str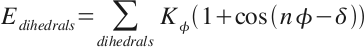
|
|
|
|