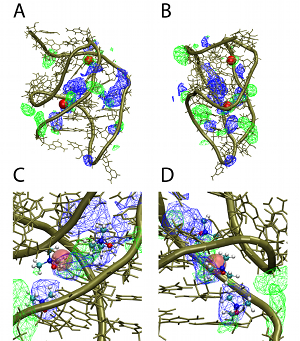
Current ResearchIntrinsic and environmental contributions to macromolecular conformational heterogeneity in DNA, RNA, carbohydrates and proteinsTowards understanding the intrinsic and environmental contributions to the conformational properties of macromolecules we apply both quantum mechanical (QM) and empirical force field methods, including enhanced sampling methods developed in our laboratory. In studies on RNA a combined QM/Bioinformatics approach was applied to understand the role of the 2'-OH group of RNA on its ability to sample a wide range of tertiary structures while sampling only A-form conformations at the secondary (duplex) structural level [1]. Results revealed that the orientation of the 2'-OH proton significantly effects the conformational flexibility of the phosphodiester backbone, such that when the proton is directed towards the base it leads to larger energy barriers, thereby stabilizing the A form of RNA duplexes. However, if it points towards the phosphodiester backbone it significantly lowers the energy barriers, thereby allowing RNA to sample a wide range of conformations at the tertiary stuctural level. Concerning environmental effects on RNA, MD simulation studies on RNA in the presence of the osmolyte TMAO revealed that the protonated form of TMAO is responsible for its ability to stabilize RNA tertiary structure [2]. This is due to protonated TMAO mimicking the behavior of positively charged ions as well as the ligand of the PreQ1 riboswitch, which was the model RNA system used in the study. In a study using the polarizable Drude-2013 the differential impact of the monovalent ions on X-ray scattering spectra was observed, representing a prediction of the force field [3], and the polarizable force field was shown to more accurately treat the ionic environment of DNA versus the additive CHARMM36 force field [4]. In the area of carbohydrates and proteins, we've developed enhanced sampling approaches in the context of Hamiltonian Replica Exchange, with emphasis on the sampling about the glycosidic linkages of complex glycans [5] as well as the phi, psi dihedrals in the peptide backbone. These approachs have been applied to trisaccharides [6] and to glycoproteins [7] and are being extended to more complex glycans associated with the Salmonella vaccine antigen and the glycans on the HIV envelope. In addition, we've used both enhanced sampling methods and standard MD simulations to investigate the folding and unfolding mechanisms of polypeptides [8,9], with the results showing the importance of the use of a force field that contains explicit electronic polarizability based on the classical Drude Oscillator model to allow for cooperative effects to be included in peptide folding and unfolding events. |
|
|
Force Field DevelopmentThe MacKerell laboratory is responsible for developing and maintaining the empirical force fields developed in the context of program CHARMM, including both additive and polarizable forms of the force fields. The CHARMM additive force field is widely used in simulations of proteins, nucleic acids, lipids, carbohydrates and small molecules, including in the use of computer-aided drug design. Beyond CHARMM, the force field may also be used in a number of simulation packages, including NAMD, GROMACS, and ChemShell QM/MM. A central focus of the lab is the developement and continued optimization of the CHARMM force field and its extension to an ever-growing library of molecules. These efforts include the continued development and extension of the CHARMM General Force Field (CGenFF) for drug-like molecules. This includes the developement of the CGenFF engine that allows for rapid atom typing and parameter assignment for drug-like molecules. We partner with ParamChem to provide a convenient web interface to the CGenFF engine for non-profit users. Commerical access to the CGenFF engine may be obtained from SilcsBio LLC. |
|
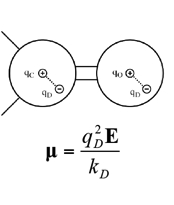
We are also developing a polarizable force field for biomolecules [1-3] based on the concept of classical Drude oscillator in collaboration with the Roux laboratory. In the Drude polarizable model, explicit polarization is introduced by attaching a charged auxiliary particle (the Drude oscillatory or particle) with a harmonic spring to the nucleus of the parent atom, which allows the atomic dipoles to adjust in response to the surrounding electronic fields. For hydrogen bond acceptors, anisotropic atomic polarizability as well as virtual particles representative of lone pairs are included in the model [3] to improve the treatment of nonbonded interactions involving the acceptors. Drude polarizable force fields for proteins [1], nucleic acids [2], water models [4,5] lipids, carbohydrates[6] and ions have been developed [7]. The Drude force field has been shown to be of utility in simulations on the microsecond time scale is undergoing extension to a wider range of molecules as well as additional optimization. Facilitating the use of the Drude force field is the implemetation of the Drude Prepper in the CHARMM-GUI. Drude Prepper converts CHARMM additive PSF and coordinate files to the corresponding Drude formatted files along with creating example inputs for MD simulations in CHARMM and NAMD. |
|
SILCS: Site-Identification by Ligand Competitive Saturation for drug design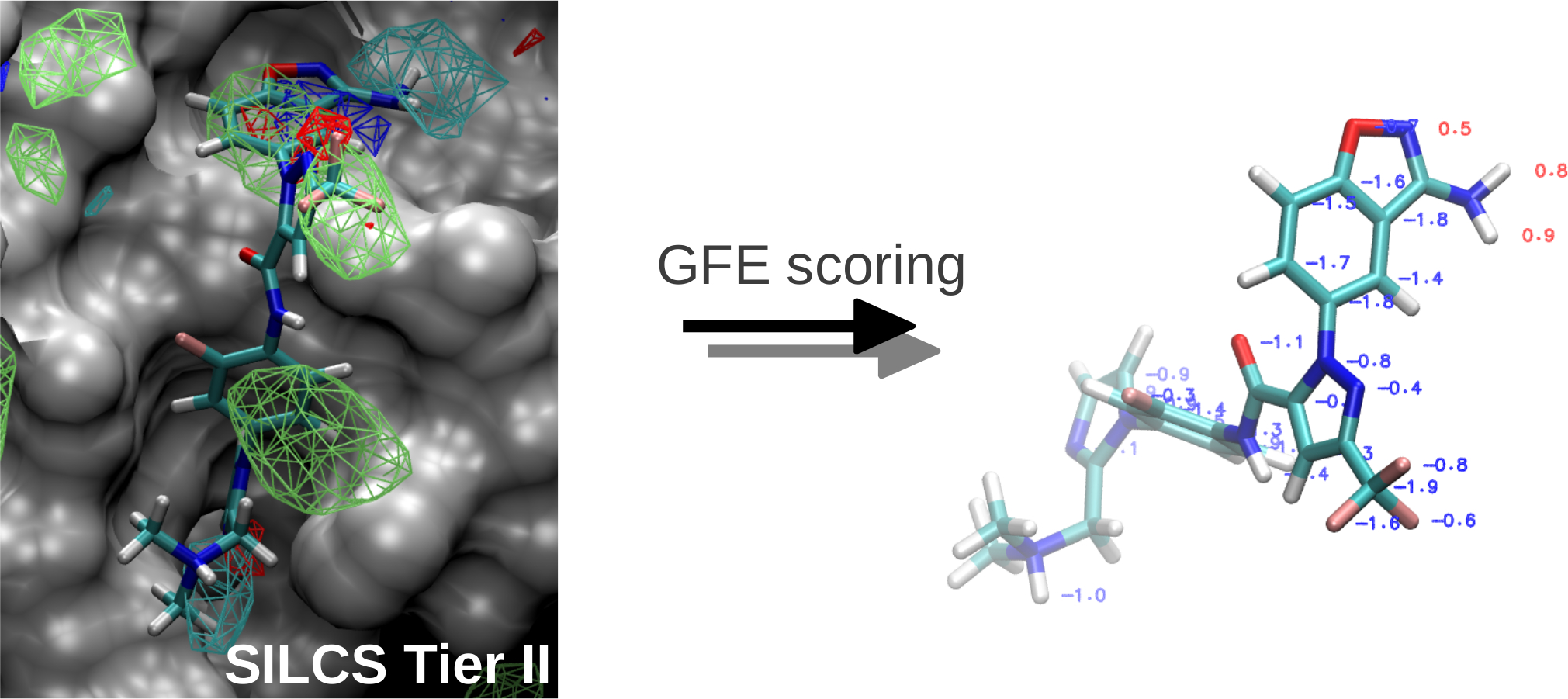
Classical molecular mechanics-based methods have been successfully applied in computer-aided drug design (CADD). However, these methods are significantly challenged by the large conformational space of flexible macromolecular targets and ligands in the aqueous environment. Additionally, the large chemical space of potential drug-like molecules to be screened against the target hinders the large scale application of accurate CADD methods. Inspired by concepts from fragment based drug design, the Site Identification by Ligand Competitive Saturation (SILCS) method takes an exploratory and modular approach to CADD. It attempts to solve the above mentioned conformational and chemical space problems simultaneously by simulating competitive binding through MD simulations to obtain the affinity pattern of target macromolecules for chemically diverse functional groups. The small molecule distributions obtained from SILCS simulations are converted to residence probability maps of fragment atoms that are then Boltzmann transformed into a free energy representation, termed grid free energy (GFE) fragment maps (FragMaps). Notably, these representations account for fragment desolvation and protein flexibility. Through our recent works, we have validated the methodology [1-6] using experimental data and application to drug design projects [7]. We have shown FragMaps to capture crystallographic interactions between proteins and ligand functional groups [1,2,5], identify cryptic binding sites [4], and have developed novel allied approaches for lead optimization [3] and pharmacophore based lead identification [6]. An important extension of the SILCS approached involved development of a novel Grand Canonial Monte Carlo approach to allow for sampling of solutes in an explicit water environment [8] allowing application of the method to systems with deep and occluded pockets such as GPCRs and nuclear receptors [9]. Current research involves further development of methodologies that make use of the rich information provided by the SILCS approach and application of the novel methods in drug design and discovery projects. The SILCS technology is currently licensed to SilcsBio LLC for commercial use. |
|

|
|
Alex explains the technique in this video. |
Conformationally Sampled PharmacophoreConformationally sampled pharmacophore (CSP) is a pharmacophore-based method that uses molecular dynamics (MD) simulations to generate an ensemble of ligand conformations from which probability distributions are calculated for select distances and angles in the molecule (i.e. pharmacophore features) [1]. Correlation of the pharmacophoric features to biological activity then allows us to identify biologically important geometric features and also to predict the activity of other ligands. In the MacKerell lab, CSP has been applied to three important classes of ligands: opioids, bile acids, and antibiotics. For the opioid receptor δ [1-3], CSP has been applied to develop a model able to distinguish agonists from antagonists, while the CSP model applied to μ receptor ligands [4] has provided a comprehensive structure-activity relationship explaining how ligand modification alters activity. This includes the use of CSP to facilitate the design of an opioid having a decreased tolerance profile. The application of CSP-SAR to bile acid transporters has yielded a model that indicates that ligands with higher intramolecular hydrogen bonding are more active against human apical sodium-dependent bile acid transporter (hASBT) due to increased hydrophobicity. And, the model also has predicted that dianionic bile acid conjugates utilize a molecular switch controlled by the location of a carboxyl group that is involved in intramolecular hydrogen bonding in order to achieve high binding affinities. For antibiotics, CSP has been applied to analogs of the antibiotic telithromycin to show that removal of methyl groups from the macrolactone ring reduces activity compared to telithromycin by increasing the conformational flexibility of the analog (see Figure) [5-7]. |
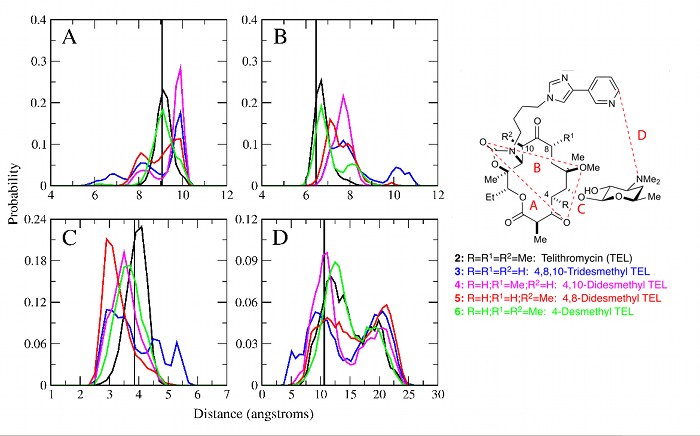
|
Computer-Aided Drug DesignUsing the SILCS and CSP methods developed in our laboratory as well as range of additional computational approaches, the MacKerell laboratory participates in a range of collaborative studies with biology and chemistry labs. These efforts involve a wide range of biological targets. Examples include efforts to develop novel antibiotics targeting a variety of systems, including the ribosome, as shown in the image below [1,2], lipid II [3], heme oxygenase and the TLR2 TIR receptor [4]. Additional efforts include the development of novel opioid analgesics with lower tolerance profiles [3] and work on a number of targets related to cancer including BCL6 [7], NOTCH [8], ERK [9], and BCL-2 family proteins [10]. |
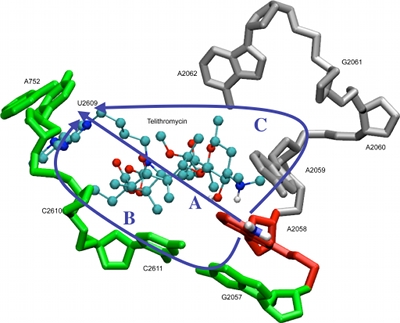
|
|
 |